Det Cornelia de Lange syndrom er en patologi af genetisk oprindelse, der er kendetegnet ved tilstedeværelsen af en signifikant kognitiv forsinkelse ledsaget af forskellige misdannende fysiske træk.
På det kliniske niveau observeres tre forskellige kliniske forløb: svær, moderat og mild. Tegn og symptomer udgøres normalt af en atypisk ansigtskonfiguration, muskuloskeletale misdannelser og forsinket kognitiv og psykomotorisk udvikling. Derudover kan andre typer af abnormiteter relateret til hjerte-, lunge- og / eller fordøjelsesdeformationer skelnes..
Med hensyn til oprindelsen af Cornelia de Lange syndrom har dets etiologi været relateret til tilstedeværelsen af specifikke mutationer i generne SMC3, SMC1A, NIPBL, blandt andre. Diagnosen er grundlæggende klinisk, baseret på fysiske og kognitive egenskaber. Imidlertid ledsages det normalt af en bekræftende genetisk test.
Behandlingen er rettet mod påvisning og behandling af medicinske komplikationer. Medicinsk, taleterapi, neuropsykologisk intervention og specialundervisning er afgørende.
Artikelindeks
Dette syndrom blev oprindeligt beskrevet af Dr. Cornelia de Lange i 1933. Hendes forskning var baseret på undersøgelsen af to patienter i alderen 6 og 17 måneder. Hans kliniske billede var præget af en alvorlig forsinkelse i fysisk vækst og intellektuel udvikling forbundet med forskellige misdannende træk..
I betragtning af ligheden mellem begge tilfælde antog den første kliniske rapport om denne patologi eksistensen af en fælles og offentlig etiologisk årsag.
Tidligere havde Brachmann (1916) formået at offentliggøre nogle obduktionsdata for en patient i alderen med nogle egenskaber, der var kompatible med Cornelia de Lange-syndromet..
På nuværende tidspunkt er det kliniske billede af dette syndrom blevet klassificeret i tre forskellige fænotyper: svær, moderat og mild..
Cornelia de Lange syndrom er en sjælden genetisk lidelse af medfødt karakter, det vil sige dens kliniske egenskaber er tydelige fra fødslen. Det defineres som værende en multisystemisk sygdom med symptomer forbundet med forsinket fysisk og kognitiv udvikling, kranio-ansigtsmisdannelser eller muskuloskeletale misdannelser..
Selvom det kliniske forløb og sværhedsgraden af dette syndrom kan variere betydeligt blandt de berørte, er det en sygdom med en høj dødelighed.
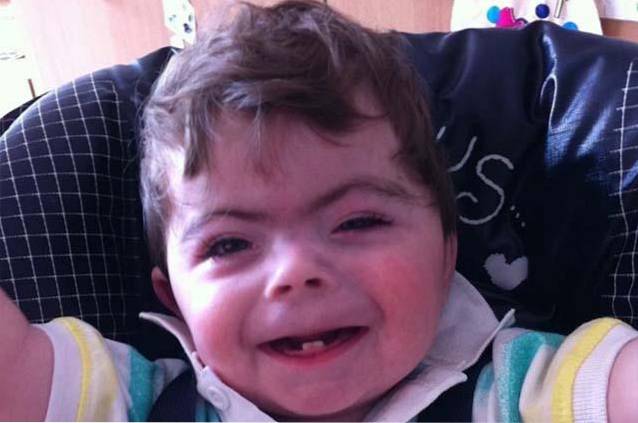
Mennesker med Cornelia de Lange syndrom er kendetegnet ved at have en atypisk eller karakteristisk ansigtskonfiguration og en forsinkelse i præ og postnatal vækst / udvikling..
Det mest almindelige er, at indlæringsproblemer vises, forsinket sprogtilegnelse eller gangart og adfærdsmæssige abnormiteter.
Cornelia de Lange syndrom er en sjælden sygdom i den almindelige befolkning, det klassificeres normalt inden for sjældne sygdomme. Epidemiologiske data er ikke ligefrem kendt. Dens forekomst er blevet estimeret til et tilfælde pr. 10.000-30.000 fødsler..
Til dato kan vi finde mere end 400 forskellige tilfælde af Cornelia de Lange syndrom beskrevet i den medicinske og eksperimentelle litteratur..
Det er en patologi, der kan påvirke begge køn i lige store antal. Nogle forfattere som Gutiérrez Fernández og Pacheco Cumani (2016) antyder en lille overvægt over for kvinder med et forhold på 1,3 / 1.
I forhold til resten af sociodemografiske faktorer har den nuværende forskning ikke identificeret en forskellig prævalens forbundet med specifikke lande eller etniske og / eller racegrupper..
En god del af de diagnosticerede tilfælde er sporadiske, selvom flere berørte familier er blevet identificeret med et klart mønster af dominerende arv..
Tegn og symptomer på Cornelia de Lange syndrom er kendetegnet ved deres brede involveringsmønster.
Denne sygdom er defineret ved tilstedeværelsen af karakteristiske ansigtsegenskaber, muskuloskeletale misdannelser i øvre og nedre lemmer, generaliseret præ- og postnatal væksthæmning sammen med udviklingen af andre fysiske abnormiteter..
Dernæst vil vi beskrive nogle af de hyppigste kliniske træk ved Cornelia de Lange syndrom:
Hos mere end 90% af dem, der er ramt af Cornelia Lange-syndrom, er det muligt at identificere en forsinkelse i fysisk udvikling eller global hypogvækst. Vækst påvirkes normalt både prænatalt og postnatalt.
De mest almindelige egenskaber hos nyfødte er:
Disse forhold varer normalt i voksenalderen. I den kan der skelnes mellem en vækst, der er under den forventede for den berørte persons køn og biologiske alder.
Sammen med denne type ændringer kan nogle abnormiteter relateret til fodring identificeres. Vanskeligheder med at sluge eller tygge mad er almindelig i de tidlige stadier af livet.
Kombinationen af kraniale og ansigtsændringer resulterer i udviklingen af en karakteristisk ansigtsfænotype hos mennesker, der lider af Cornelia de Lange syndrom..
Nogle af de mere almindelige abnormiteter inkluderer:
Forsinkelse i kognitiv og psykomotorisk udvikling er en af de centrale kliniske fund i Cornelia Lange syndrom. En langsom tilegnelse af færdigheder relateret til motorisk eller mental aktivitet identificeres normalt.
De mest berørte milepæle er erhvervelse af siddende, affektivt smil, pludrende, uafhængig bevægelse, udsendelse af de første ord, forståelse og ordrer, fodring, ambulation eller uafhængigt toilet.
Hos mange af de berørte kan der identificeres en gennemsnitlig IQ forbundet med mild eller moderat intellektuel handicap.
Adfærd hos dem, der er ramt af Cornelia de Lange syndrom, præsenterer normalt nogle særpræg:
Cornelia de Lange syndrom er også forbundet med udviklingen af forskellige medicinske komplikationer.
De hyppigste dødsårsager eller forværring af de berørte medicinske status er relateret til:
Variationen af tegn og symptomer på Cornelia de Lange syndrom har gjort det muligt at klassificere dets kliniske forløb:
Det er normalt det mest alvorlige. Ændringerne og anomalierne er kendetegnet ved tilstedeværelsen af fysisk underskov, muskuloskeletale misdannelser, unormale ansigtsegenskaber, begrænsning af ledmobilitet, kognitiv forsinkelse og andre medicinske komplikationer (auditiv, okulær, fordøjelsessystem, reno-urologisk, hjerte- og kønsorgan)..
I denne undertype er de fysiske ændringer normalt ikke særlig tydelige, især i ekstremiteterne. De berørte har normalt ikke alvorlige intellektuelle underskud. Det mest almindelige er, at diagnosen stilles ud over det nyfødte stadium.
Dets kliniske forløb er grundlæggende præget af klinisk variation. Ansigtsegenskaber er til stede i de fleste tilfælde, men udtrykket for resten af anomalierne er variabelt..
Oprindelsen af Cornelia Lange syndrom er forbundet med tilstedeværelsen af genetiske abnormiteter. I de undersøgte tilfælde var det muligt at identificere specifikke mutationer i 5 forskellige gener: NIPBL, SMC1A, HDAC8, RAD21 og SMC3.
Den mest almindelige ændring er relateret til NIPBL-genet, identificeret hos mere end halvdelen af de berørte. Resten af genetiske anomalier er mindre hyppige.
Alle disse gener spiller en fremtrædende rolle i produktionen af proteiner relateret til cohesin-komplekset, der er ansvarlig for reguleringen af kromosomstruktur og organisering, stabilisering af genetisk information i celler og DNA-reparation..
Derudover udfører de også flere grundlæggende funktioner i den prænatale udvikling af ekstremiteterne, ansigtet og andre regioner og kropssystemer.
Diagnosen af Cornelia de Lange syndrom er klinisk. I øjeblikket er der ingen laboratorietest, der definitivt indikerer dets tilstedeværelse. Inden for det medicinske område er det mest almindelige at bruge de diagnostiske kriterier foreslået af Kline et al..
Disse refererer til identifikation af kraniofaciale anomalier, i vækst og udvikling, i ekstremiteterne, neurosensoriske og kutane ændringer, adfærdsmæssige lidelser osv..
Derudover er det vigtigt at udføre en molekylær genetisk analyse for at identificere tilstedeværelsen af mutationer forbundet med Cornelia de Lange syndrom..
Selvom der ikke er nogen kur mod Cornelia de Lange-syndromet, involverer dets terapeutiske tilgang design af en kontinuerlig medicinsk opfølgning sammen med behandling af komplikationer..
Forfatterne Gil, Ribate og Ramos (2010) påpeger nogle af de mest anvendte tilgange.



Endnu ingen kommentarer