Det Canavan sygdom Det er en sjælden genetisk sygdom, der opstår, fordi nerveceller i hjernen er beskadiget og ude af stand til at kommunikere med hinanden. Denne sygdom er til stede i ethvert samfund og etnisk gruppe, selvom den er meget hyppigere i den jødiske befolkning i Ashkenazi og deres efterkommere, hvor 1 ud af 6.400-13.000 mennesker er ramt. Den globale prævalens er ukendt.
Denne sygdom er inden for gruppen af leukodystrofi. Denne kategori inkluderer alle genetiske lidelser, hvor myelinskeden, der omgiver neuroner axoner, er beskadiget, og der er derfor ingen god kommunikation mellem neuroner.
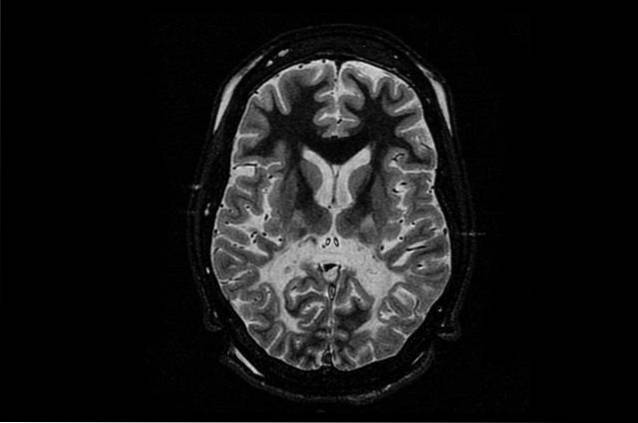
Den mest almindelige og samtidig mest alvorlige form for denne sygdom er neonatal eller infantil. Denne form for Canavan-sygdom rammer nyfødte børn eller i deres første leveår..
Børn, der lider af denne sygdom, udgør ingen problemer i de første måneder af livet, men disse begynder at blomstre mellem 3 og 5 måneder. De vigtigste symptomer skyldes udviklingsunderskuddet, hvor børn har motoriske problemer, der forhindrer dem i at dreje rundt, dreje hovedet eller sidde uden nogen støtte.
Andre almindelige symptomer er muskelsvaghed (hypotoni), unormal hovedudvikling (makrocephali) og irritabilitet. I mindre grad kan de også have problemer med at spise, krampeanfald og søvnproblemer..
En anden mindre almindelig form er Canavan sygdom, der begynder i mellembarndommen eller ungdommen. Børn og unge med denne sygdom har problemer med sprogudvikling og motoriske færdigheder, men disse problemer er ofte så milde, at de ikke identificeres som symptomer på Canavan-sygdommen..
Forventet levetid for mennesker med Canavan-sygdom er meget heterogen og varierer især afhængigt af tidspunktet for sygdommens begyndelse..
Børn, der lider af neonatal eller infantil form, lever normalt kun få år, selvom nogle når ungdomsårene og meget få indtil voksenalderen. Mens de, der lider af den unge form, har en normal forventet levetid.
Artikelindeks
Der er to veldifferentierede former for Canavan-sygdom: den af nyfødt eller infantil debut og den af debut i mellembarndommen eller ungdomsårene..
Symptomer på nyfødt eller barndomsudbrud af Canavan er meget alvorlige, normalt ikke mærkbare indtil 3-50 måneders alderen og inkluderer makrocephali, tab af motorisk kontrol af hovedet og udviklingsunderskud. Udviklingsunderskud bliver tydeligere, når barnet bliver ældre.
De mest alvorlige symptomer er dem, der er relateret til motoriske problemer, da børn ikke er i stand til at sidde eller stå op uden støtte, gå eller tale. Når de bliver ældre, kan hypotoni føre til spasticitet.
Selvom de har alle disse motoriske problemer, kan de lære at interagere socialt, smile, pege på objekter ...
Nogle børn lider også af optisk atrofi, hvilket forårsager visuelle problemer, selvom de stadig kan identificere objekter visuelt.
Efterhånden som symptomerne vokser, bliver de værre og forårsager søvnbesvær, kramper og fodringsproblemer. Barnet bliver totalt afhængigt og har brug for hjælp til at udføre enhver opgave.
Forventet levetid for disse børn er ganske kort, de fleste dør om få år, selv om nogle lever indtil ungdomsårene eller voksenalderen.
Canavan sygdom med debut i middelbarndommen eller ungdommen er mildere end den forrige. Symptomer inkluderer nogle vanskeligheder i verbal og motorisk udvikling.
Selvom de normalt er så milde, at de ikke identificeres som symptomer på Canavan-sygdommen, diagnosticeres denne sygdom normalt efter en urinanalyse, da en af markørerne er den høje koncentration af N-acetyl asparaginsyre (NAA i urinen.
Denne sygdom er forårsaget af en mutation i et gen kaldet ASPA. Dette gen er det, der styrer enzymet aspartoacylase, som er ansvarlig for nedbrydende NAA-molekyler..
Mutationen af ASPA-genet får aspartoacylase til at reducere dets effektivitet, så det nedbrydes ikke nok NAA-molekyler, og der vil være en høj koncentration af dette stof. Jo tidligere denne mutation opstår, jo dårligere virkninger har den.
Selvom funktionen af NAA-molekyler ikke er særlig godt forstået, ser det ud til, at de er involveret i transporten af vandmolekyler gennem neuroner, og overskuddet af dette stof forhindrer dannelse af nyt myelin og ødelægger det eksisterende. Dette medfører, at forbindelserne mellem neuroner ikke fungerer korrekt, og hjernen ikke er i stand til at udvikle sig normalt..
Desuden kan denne sygdom arves på en autosomal recessiv måde. Så hvis hvert medlem af parret er bærer af den patogene variant af ASPA-genet, og de beslutter at få et barn, vil de sandsynligvis:
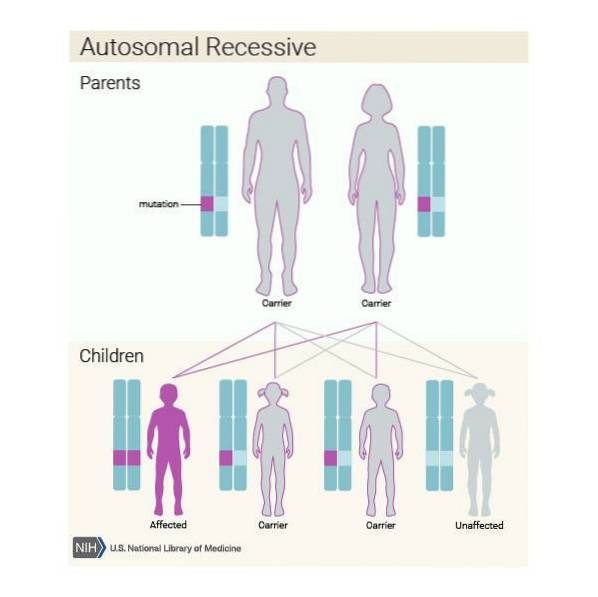
Det er meget vigtigt, at individer, der tilhører risikopopulationen, i dette tilfælde efterkommere af Ashkenazi-jøder, gennemgår en genetisk analyse for at kontrollere, om de bærer ASPA-genet, før de får et barn..
Behandlingen afhænger af sygdomsformen og de symptomer, som hver enkelt præsenterer..
Der er i øjeblikket ingen kur mod Canavan-sygdommen, så tilgængelige terapier fokuserer på at forbedre patientens livskvalitet ved at støtte, pleje og hydrere og forebygge og behandle infektioner.
Det anbefales, at børn får fysioterapeutisk behandling for at forbedre deres kropsholdning og motoriske færdigheder, for at undgå og behandle kontrakturer og muskelproblemer, såsom tryksår. De kan også deltage i terapeutiske og uddannelsesmæssige programmer for at forbedre deres kommunikationsevner..
Behandling med medicin inkluderer antiepileptiske lægemidler (AED'er), hvis barnet har anfald, acetazolamid (mærke Diamox®) for at reducere intrakranielt tryk og injektioner af botulinumtoksin (Botox®) til behandling af spasticitet, hvis den er til stede.
Det er nødvendigt at foretage en opfølgning hver sjette måned for at kontrollere, hvilken tilstand barnet er i, og hvordan dets udvikling går.
Mennesker, der lider af denne form for sygdommen, oplever meget mildere symptomer, så de har normalt kun brug for terapier for at forbedre deres sprog eller specielle uddannelsesprogrammer. De har ikke brug for medicin.
Årlig overvågning af barnets tilstand anbefales.
Effekten af andre terapier undersøges i øjeblikket i både mennesker og dyremodeller..
Effektiviteten af en genetisk transplantation til hjernen hos børn med Canavan-sygdom undersøges ved hjælp af en ikke-viral vektor.
De første resultater viser, at denne type transplantation tolereres godt af børn og forårsager nogle biokemiske, radiologiske og metaboliske ændringer, men det er ikke nyttigt til helbredelse af sygdommen, så der udføres stadig test (Leone et al 2000, Janson et al. . til 2002).
McPhee et al. (2006) gennemfører en undersøgelse, hvor det sunde ASPA-gen transplanteres til forskellige steder i børnenes krop ved hjælp af AAV2 som en vektor. I en af testene, hvor 10 frivillige børn deltog. Hos 3 af dem arbejdede transplantationen og neutraliserede deres antistoffer, men ingen af børnene forbedrede sig.
Lithiumcitrat kan reducere niveauet af NAA-koncentration i hjernen, hvorfor Assadi et al. (2010) besluttede at gennemføre et eksperiment, hvor de administrerede lithiumcitrat til 6 personer med Canavan-sygdom i 60 dage.
Der blev fundet niveauer af NAA i basalganglier og hvidt stof i frontallappen, selvom der ikke blev fundet kliniske forbedringer.
Manglen på aspartoacylaseenzymer forårsager lave niveauer af acetat i hjernen, så Mahavarao og hans team (2009) besluttede at give glyceroltriacetat til to patienter med Canaval's sygdom for at hæve deres acetatniveauer og se om det også øgede aspartoacylase-niveauet.
Forbindelsen tolereredes godt af patienterne, selvom der ikke blev fundet kliniske forbedringer. De gennemfører i øjeblikket tests, der administrerer en større mængde glyceroltriacetat.
En af måderne til at skabe dyremodeller, der repræsenterer en sygdom, er at skabe dyr slå ud. Disse dyr, normalt mus, er genetisk modificerede til at fjerne eller ændre genet, der er ændret i sygdommen. I dette tilfælde er det modificerede gen ASPA-genet..
Dyremodeller bruges til bedre at forstå sygdommen, studere dens biologiske korrelat og kontrollere effektiviteten af nye behandlinger.
Matalon et al. (2003) brugte mus slå ud for at teste effektiviteten af en genterapi med AAV2 som en vektor. De fandt ud af, at der havde været forbedringer i myelinskederne, men kun i nogle dele, ikke hele hjernen.
Surendrans team i samarbejde med Genzyme Corporation (2004) testede en stamcelletransplantationsbehandling. De fandt ud af, at der var produceret nye oligodendrocytter, men ikke nok til at gendanne alle myelinskeder..
Et andet team testede en terapi, der bestod i at erstatte de funktionsfejlede asparthoacyclaseenzymer med nye, der blev injiceret i musens bukhinde. slå ud.
De kortsigtede resultater viste, at enzymerne var i stand til at passere blod-hjerne-barrieren (nåede deres mål) og var i stand til at reducere niveauerne af NAA i hjernen signifikant. Selv om disse resultater er lovende, er en langsgående undersøgelse nødvendig for at verificere de langsigtede effekter (Zano et al., 2011).
De første tegn, der advarer læger om, at der er noget galt, er de fysiske, især hypotoni og makrocephali.
Normalt udføres en neuroimaging-undersøgelse normalt hos barnet for at kontrollere tegn på leukodystrofi, såsom en lavere tæthed af hvidt stof, hvis disse tegn observeres. Det skal bemærkes, at denne test er mindre effektiv hos børn med Canavan-sygdom, der begynder i midten af barndommen eller ungdommen..
Når det er bevist, at barnet lider af en leukodystrofi, udføres der mere specifikke tests for at udelukke andre sygdomme, herunder:
Det sidste trin til at bekræfte sygdommen ville være at udføre en genetisk undersøgelse som følger:



Endnu ingen kommentarer