
Det Apert syndrom eller acrocephalosyndactyly type I (ACS1) er en patologi af genetisk oprindelse, der er kendetegnet ved tilstedeværelsen af forskellige ændringer og misdannelser i kraniet, ansigtet og ekstremiteterne.
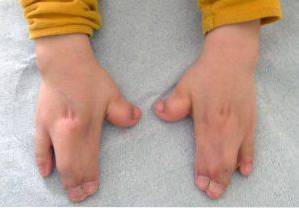
På et klinisk niveau er Apert syndrom kendetegnet ved tilstedeværelsen eller udviklingen af en spids eller langstrakt kranium, nedsænket ansigtsområde med en ændring i tændernes fremspring, fusion og lukning af fingerben og led, mental retardation variabel, sprogforstyrrelser , etc..
Selvom denne patologi kan være arvelig, forekommer Apert syndrom i de fleste tilfælde uden tilstedeværelse af en familiehistorie, hovedsageligt på grund af en de novo-mutation under drægtighedsfasen..
De genetiske mekanismer, der forårsager Apert syndrom, er ikke ligefrem kendt. I øjeblikket er forskellige genetiske ændringer, der er i stand til at producere denne patologi, blevet identificeret, hovedsagelig relateret til mutationer i FGFR2-genet.
På den anden side begynder diagnosen af Apert syndrom normalt med klinisk mistanke i den prænatale periode efter identifikationen af abnormiteter i rutinemæssige ultralydsscanninger og bekræftes ved at udføre en genetisk undersøgelse.
Med hensyn til behandling er der ingen form for helbredende intervention for Apert syndrom. Gennem historien om denne patologi er der imidlertid designet forskellige specifikke interventioner, der normalt inkluderer neurokirurgi, kraniofacial kirurgi, maxillofacial kirurgi, medikamentel behandling, fysioterapi, psykologisk og neuropsykologisk intervention.
Artikelindeks
Apert syndrom er en genetisk patologi karakteriseret ved tilstedeværelsen af forskellige skeletmisdannelser på kranie-, ansigts- og / eller lemmerniveau.
Den væsentlige ændring af Apert syndrom udgøres af en for tidlig eller tidlig lukning af kraniesprækkene, som forårsager en unormal vækst i resten af ansigtets og kraniets strukturer. Ud over disse kan misdannelser også forekomme i øvre og nedre ekstremiteter, såsom fusion af fingre og tæer..
På den anden side kan de kognitive evner hos mennesker, der lider af Apert syndrom, også blive påvirket med en variabel sværhedsgrad fra mild til moderat.
Selvom Baumgartner (1842) og Wheaton (1894) nævnte de første om denne medicinske tilstand, var det først i 1906, da den franske medicinske specialist Eugene Apert, nøjagtigt beskrev dette syndrom og offentliggjorde den første kliniske rapport..
I sin publikation beskriver Eugene Apert et sæt nye tilfælde af patienter, der er ramt af et veldefineret misdannelsesmønster og er kendetegnet ved de karakteristiske tegn og symptomer på denne patologi..
Det var således først i 1995, at de etiologiske genetiske faktorer ved Apert syndrom blev identificeret. Specifikt beskrev Wilkie et al. Tilstedeværelsen af to mutationer i FGFR2-genet hos omkring 40 berørte patienter..
Derudover er Apert syndrom en medicinsk tilstand, der er klassificeret inden for de sygdomme eller patologier, der er karakteriseret ved at præsentere kraniosynostose (for tidlig lukning af kraniale suturer).
Andre patologier, der tilhører denne gruppe, er Pfeiffer syndrom, Crouzon syndrom, Saethre-Chotzcen syndrom og Carpenter syndrom..
Apert syndrom betragtes som en sjælden eller sjælden patologi, det vil sige, det har en prævalens på mindre end et tilfælde pr. 15.000 indbyggere i den generelle befolkning..
Specifikt forekommer Apert syndrom omkring en person for hver 160.000-200.000 fødsler, og derudover er der 50% sandsynlighed for at overføre denne patologi på arveligt niveau.
Derudover er der ikke identificeret en højere forekomst hos mænd eller kvinder med hensyn til fordeling efter køn, og den har heller ikke været forbundet med bestemte etniske grupper eller geografiske placeringer..
I øjeblikket og i betragtning af at Apert syndrom blev identificeret i ca. 1984, i kliniske rapporter og i den medicinske litteratur, der har offentliggjort mere end 300 tilfælde af denne patologi.
De kliniske manifestationer af Apert syndrom inkluderer normalt en misdannelse eller ufuldstændig udvikling af kraniestrukturen, en atypisk fænotype eller ansigtsmønster og skeletændringer i ekstremiteterne..
I tilfælde af Apert syndrom er den centrale involvering relateret til dannelsen og lukningen af kraniets knoglestruktur. Under embryonal udvikling opstår en proces kaldet creneosynostose, der er kendetegnet ved for tidlig lukning af kraniale suturer.
Kraniale revner eller suturer er en type fibrøse vævsbånd, der har det grundlæggende mål at forbinde de knogler, der udgør kraniet (frontal, occipital, parietal og temporal).
I graviditetsfasen og den tidlige postnatale periode holdes knoglestrukturen, der udgør kraniet, sammen takket være disse fibrøse og elastiske væv.
Normalt kraniale knogler smelter først omkring 12 til 18 måneder. Tilstedeværelsen af bløde pletter eller mellemrum mellem kranialbenene er en del af den normale børns udvikling.
I løbet af hele barndomsfasen tillader disse suturer eller fleksible regioner hjernen at vokse på en accelereret måde og derudover beskytte den mod stød.
I Apert syndrom gør for tidlig lukning af disse kraniale suturer og kraniale knogler således normal udvikling af kranie- og hjernevækst umulig..
Derfor kan de mest almindelige tegn og symptomer på Apert syndrom omfatte:
Disse typer af ændringer påvirker hovedsageligt de øvre og nedre ekstremiteter, normalt fusion og udvikling af fingrene..
Ud over disse er det også muligt at observere andre kliniske fund på muskuloskeletalt niveau, forkortelse af forskellige knogler (radius, humerus, lårben), hypoplasi i scapula eller bækken, fusion af livmoderhvirvler.
Som en konsekvens vil mange berørte præsentere nedsat ledmobilitet og kan derfor udvikle forskellige vanskeligheder til erhvervelse af grov og finmotorik.
Disse typer af anomalier er meget heterogene og varierer blandt berørte individer, men nogle af de mest almindelige er blevet identificeret:
Den etiologiske ændring af denne patologi kan føre til udvikling af læsioner eller sekundære patologier på et morfologisk og strukturelt niveau i forskellige områder af kroppen, nogle af dem inkluderer:
På trods af at det i mange tilfælde er muligt at observere tilstedeværelsen af en generel ændring af kognitive funktioner og intellektuelt niveau, er mental retardation ikke utvetydigt til stede i alle tilfælde af Apert syndrom..
Derudover kan dette i tilfælde, hvor der er en forringelse af det intellektuelle niveau, variere på en skala fra mild til moderat.
På det sproglige område er udviklingen af forskellige underskud hyppig, hovedsageligt relateret til artikulation af lyde som et resultat af mandibulære og orale misdannelser..
Apert syndrom skyldes tilstedeværelsen af en specifik mutation i FGFR2 genet. Eksperimentelle undersøgelser har vist, at dette gen er ansvarlig for produktionen af et protein, kaldet fibroblastvækstfaktorreceptor 2.
Blandt funktionerne i denne faktor beskriver den udsendelsen af forskellige kemiske signaler til umodne celler for at forårsage deres transformation og differentiering til knogleceller under fosterets eller prænatal udviklingsfase..
Derfor tilstedeværelsen af mutationer i FGFR2 genet ændrer funktionen af dette protein og kan derfor forårsage en tidlig fusion af knoglerne i kraniet, hånden og fødderne..
En god del af de kliniske træk ved Apert syndrom kan identificeres under graviditet, specielt i ultralydsundersøgelser af graviditet og fosterudvikling..
Således, når der er en klinisk mistanke, genstartes en genetisk undersøgelse for at identificere tilstedeværelsen af en genetisk mutation, der er kompatibel med Apert syndrom..
På den anden side, når tegnene er subtile eller ikke er blevet identificeret inden fødslen, er det efter dette muligt at udføre en detaljeret fysisk analyse og forskellige genetiske tests for at bekræfte diagnosen..
Selvom der ikke er nogen specifik kur mod Apert syndrom, er der beskrevet forskellige tilgange til behandling af symptomer og medicinske komplikationer ved denne patologi..
De mest effektive terapeutiske indgreb er dem, der implementeres tidligt i de første øjeblikke i livet og involverer fagfolk fra forskellige områder.
Behandlingen af berørte børn kræver typisk individuel planlægning med flere operationer planlagt. Således er styringen af denne patologi baseret på korrektion af skelet- og kranio-ansigtsmisdannelser og psykologisk og neuropsykologisk støtte..
Gennem neurokirurgi er målet at rekonstruere kranievælvet, mens specialister inden for maxillofacial kirurgi forsøger at korrigere misdannelser i ansigtet. På den anden side er traumakirurgers deltagelse også hyppig til rekonstruktion af misdannelser i hænder og fødder.
Derudover er designet af individualiserede programmer til tidlig stimulering, kommunikationsrehabilitering, træning af sociale færdigheder eller psyko-pædagogisk opfølgning gavnlig for at opnå en optimal, funktionel og uafhængig udvikling af de berørte personer..


Endnu ingen kommentarer