Det kraniosynostose Det er et sjældent problem med kraniet, der får barnet til at udvikle eller præsentere deformationer i hovedet ved fødslen. Det handler om den tidlige fusion af de forskellige dele af kraniet, så det ikke kan vokse ordentligt og afbryder den normale udvikling af både hjernen og kraniet..
Hos den nyfødte består kraniet af flere knogler, der endnu ikke er sammenføjet, dette er således, at hjernen har plads nok til, at den kan fortsætte med at udvikle sig. Faktisk vil kraniets knogler fordoble sig i løbet af de første tre måneder af livet og smelter ikke helt sammen i slutningen af teenagere..

I virkeligheden ændres niveauet af fusion af kraniet knogler med alderen og afhængigt af suturerne; lukke nogle før andre. Den nyfødte kranium består af syv knogler, og de udvikler sig gennem to processer: knoglediagnosticering og knoglemodellering..
Det ser ud til, at kraniet består af et enkelt kompakt stykke, men i modsætning til hvad du måske tror, er kraniet mere som en fodbold: den har en række knogler arrangeret i plader, der passer sammen og bygger en kugle..
Mellem disse plader er stærke elastiske væv kaldet suturer. Det er det, der giver kraniet fleksibilitet til at udvikle sig, når hjernen vokser. Denne fleksibilitet gør det også muligt for barnet at blive leveret gennem fødselskanalen..
Hvad der sker er, at når et område af kraniet, der vokser, smelter sammen og lukker, vil andre områder forsøge at kompensere for dette, blive mere fremtrædende og ændre hovedets normale form..
Kraniosynostose kan også forekomme i litteraturen som synostose eller for tidlig lukning af suturer.
Artikelindeks
Der er flere typer kraniosynostose afhængigt af de dele af kraniet, der ændres og den resulterende form af hovedet..
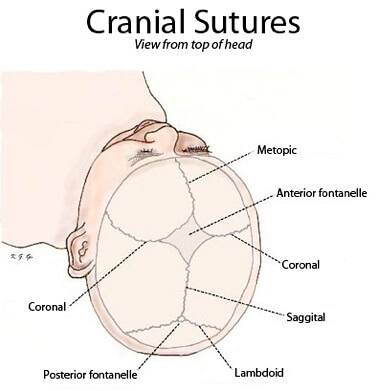
Det er den mest almindelige type og rammer mænd oftest. Det er den for tidlige sammensmeltning af den sagittale sutur, som er placeret i midterlinjen af den øverste del af kraniet og går fra det bløde punkt (også kaldet fontanel) til bagsiden af hovedet.
Det giver anledning til en lang og smal hovedform. Når den vokser, bliver bagsiden af hovedet mere fremtrædende og spids, og panden stikker ud. Denne type er den, der udgør det mindste problem for normal hjerneudvikling og er relativt let at diagnosticere.
Den består af den tidlige forening af en af de koronale suturer, hvor hjernens pande og frontlobe vokser fremad. I denne type ser panden fladt ud, og øjenlågene er hævet og skrånende.
Derudover stikker disse ud, og næsen afviges også til den ene side. Et tegn på, at babyen har denne type kraniosynostose, er at han vil vippe hovedet til den ene side for at undgå at se dobbelt..
Det er krydset mellem metopisk sutur, som er placeret i midten af panden på den berørte person og går fra det bløde punkt eller fontanelle til begyndelsen af næsen.
Det får øjenstikkene til at komme sammen, og øjnene er meget tæt på hinanden. Panden har en fremspringende trekantet form.
Det er det mindst almindelige og skyldes en for tidlig lukning af en af de lammede suturer, der er bag på hovedet.
Dette resulterer i en udfladning af dette område af kraniet, hvilket får knoglen bag øret (mastoidbenet) til at stikke ud, hvilket manifesteres i det ene øre lavere end det andet. I dette tilfælde kan kraniet også vippes til den ene side..
Alle disse svarer til fagforeninger i en enkelt sutur, men der kan også være foreninger på mere end en.
For eksempel:
- Forreste brachycephaly: det kaldes bicoronal, som det sker, når de to koronale suturer, der går fra øre til øre, smelter sammen for tidligt og præsenterer panden og øjenbrynområdet fladt. Kraniet ser generelt bredere ud end normalt.
- Bageste brachycephaly: kraniet er også udvidet, men på grund af foreningen af de begge lammede suturer (som, som vi sagde, er i ryggen).
- Scaphocephaly af satigal sutur og metopic: hovedet har et aflangt og indsnævret udseende.
For eksempel:
- Turribrachycephaly, ved forening af bikoronal, sagittal og metopisk sutur: hovedet er spids og er karakteristisk for Apert syndrom.
- Multisuturer sammen, der giver kraniet en "kløverblad" -form.
Denne sygdom er sjælden og rammer omkring 1 ud af 1.800 til 3.000 børn. Det er mere almindeligt hos mænd, hvor 3 ud af 4 mandlige tilfælde er berørt, selvom dette ser ud til at variere afhængigt af typen kraniosynostose..
Mellem 80% og 95% af tilfældene tilhører den ikke-syndromiske form, det vil sige som en isoleret tilstand, skønt andre forfattere har anslået, at mellem 15% og 40% af patienterne kan være en del af et andet syndrom.
Med hensyn til typer kraniosynostose ser det ud til, at den hyppigste er den, der involverer den sagittale sutur (40-60% af tilfældene) efterfulgt af koronale (20-30%) og derefter metopikken (10% eller mindre ). Foreningen af lambdoid-suturen er meget sjælden.
Kraniosynostose kan være syndromisk, dvs. det er knyttet til andre sjældne syndromer. Et syndrom er en række associerede symptomer, der opstår af samme årsag, og som i de fleste tilfælde normalt er genetiske.
Hvad der er kendt som sekundær kraniosynostose kan også forekomme, hvilket er mere almindeligt, og som har sin oprindelse i en svigt i hjernens udvikling.
Normalt vil det føre til mikrocefali eller reduceret kraniestørrelse. Derfor vil craniosynostose her vises sammen med andre problemer med hjernens udvikling, såsom holoprosencephaly eller encephalocele..
Det kan også være forårsaget af teratogene midler, det betyder ethvert kemisk stof, mangel eller noget skadeligt fysisk middel, der frembringer morfologiske ændringer i fosterstadiet. Nogle eksempler er blandt andet stoffer som aminopterin, valproat, fluconazol eller cyclophosphamid..
- På den anden side kan det også være ikke-syndromisk; årsagerne er variable og ikke fuldt kendte.
Det er kendt, at der er faktorer, der kan lette kraniosynostose, såsom:
- Lille plads i livmoderen eller unormal form af livmoderen, hvilket hovedsagelig forårsager koronal synostose.
- Forstyrrelser, der påvirker knoglemetabolismen: hyperkalcæmi eller rakitis.
- Det kan undertiden skyldes hæmatologiske (blod) lidelser, såsom medfødt hæmolytisk gulsot, seglcelleanæmi eller thalassæmi.
- I nogle tilfælde er kraniosynostose en konsekvens af iatrogene problemer (dvs. forårsaget af fejl fra en læge eller sundhedspersonale)
I nogle tilfælde kan craniosynostose måske ikke mærkes før et par måneder efter fødslen. Specifikt, når det er forbundet med andre kraniofaciale problemer, kan det ses fra fødslen, men hvis det er mildere eller har andre årsager, vil det blive observeret, når barnet vokser.
Derudover vises nogle symptomer beskrevet her normalt i barndommen..
- Den grundlæggende er kraniets uregelmæssige form, der bestemmes af den type kraniosynostose, den har..
- En hård ryg kan mærkes ved berøring ved suturkrydsningsområdet.
- Den bløde del af kraniet (fontanel) kan ikke mærkes eller er anderledes end normalt.
- Babyens hoved ser ikke ud til at vokse i forhold til resten af kroppen.
- Der kan være en stigning i intrakranielt tryk, der kan forekomme i enhver form for kraniosynostose. Det skyldes åbenbart misdannelser i kraniet, og jo flere suturer der er forbundet, jo mere almindelig vil denne stigning være, og jo mere alvorlig er den. F.eks. Vil der i typen af flere suturer forekomme en stigning i intrakranielt tryk i ca. 60% af tilfældene, mens procenten falder til 15% i tilfælde af en enkelt sutur..
Som en konsekvens af det foregående punkt vil følgende symptomer også forekomme i kraniosynostose:
- Vedvarende hovedpine, hovedsageligt om morgenen og om natten.
- Synsvanskeligheder som at se dobbelt eller sløret.
- Hos lidt ældre børn faldt den akademiske præstation.
- Forsinket neuroudvikling.
- Hvis øget intrakranielt tryk ikke behandles, kan opkastning, irritabilitet, langsom reaktion, hævelse af øjnene, problemer med at følge et objekt med syn, høre- og åndedrætsproblemer opstå.
Det er vigtigt, at ikke alle kraniumdeformiteter er kraniosynostose. For eksempel kan en unormal hovedform opstå, hvis babyen forbliver i samme position i lang tid, såsom at ligge på ryggen..
Under alle omstændigheder er det nødvendigt at konsultere en læge, hvis det observeres, at babyens hoved ikke udvikler sig ordentligt eller har uregelmæssigheder. Diagnosen foretrækkes imidlertid, da rutinemæssige pædiatriske kontrolbesøg foretages til alle babyer, hvor specialisten undersøger kraniets vækst..
Hvis det er en mildere form, kan det muligvis ikke opdages, før barnet bliver ældre, og der er stigninger i intrakranielt tryk. Derfor bør de ovennævnte symptomer ikke ignoreres, hvilket i dette tilfælde synes mellem 4 og 8 år..
Diagnosen skal omfatte:
- En fysisk eksamen: palpering af hovedet på den berørte person for at kontrollere, om der er kamme i suturerne eller for at observere, om der er ansigtsdeformiteter.
- Billedstudier, såsom computertomografi (CT), som giver dig mulighed for at se de suturer, der er vedhæftet. De kan identificeres, og det vil blive observeret, at hvor der skal være en sutur, er der ikke, ellers, at linjen stikker ud i en kam.
- Røntgenstråler: for at opnå nøjagtige målinger af kraniet (via cephalometri).
- Genetisk testning: hvis det mistænkes for, at det kan være af en arvelig type, der er knyttet til et syndrom, med det formål at opdage hvilket syndrom der ville være og behandle det så hurtigt som muligt. De kræver normalt en blodprøve, selvom der undertiden også kan testes prøver af andet væv såsom hud, celler fra kindens inderside eller hår..
Der er mere end 180 forskellige syndromer, der kan forårsage kraniosynostose, selvom de alle er meget sjældne. Nogle af de mest karakteristiske er:
- Crouzon syndrom: dette er den mest almindelige og er forbundet med bilateral koronal kraniosynostose, abnormiteter i mellemfladen og udbulende øjne. Det ser ud til at være på grund af en mutation i FGFR2-genet, selvom nogle tilfælde opstår spontant.
- Apert syndrom: Han har også bilateral koronal kraniosynostose, selvom andre former for synostose kan ses. Der er fusioner i bunden af kraniet såvel som deformiteter i hænder, albuer, hofter og knæ. Dens oprindelse er arvelig og giver anledning til karakteristiske ansigtsegenskaber.
- Tømrer syndrom: Dette syndrom er normalt forbundet med posterior plagiocephaly eller sammenslutning af lamboide suturer, selvom scaphocephaly også forekommer. Det skelnes blandt andet ved deformationer i ekstremiteterne og med et ciffer mere i fødderne.
- Pfeiffer syndrom: Unicoronal craniosynostosis er almindelig i denne tilstand såvel som ansigtsdeformiteter, der forårsager høreproblemer og i ekstremiteterne. Det er også forbundet med hydrocephalus.
- Saethre-Chotzen syndrom: de præsenteres normalt med ensidig koronal craniosynostose med meget begrænset udvikling af den forreste kraniale base, meget lav hårvækst, ansigtsasymmetri og udviklingsforsinkelse. Det er også af den medfødte type.
Der skal gøres en indsats for at udvikle tidlig behandling, da mange af problemerne kan løses ved den hurtige vækst i hjernen og fleksibiliteten i babyens kranium til at tilpasse sig ændringer..
Selv i meget milde tilfælde kan ingen specifik behandling anbefales, men det forventes, at dens æstetiske virkninger ikke vil være så alvorlige, som den berørte person udvikler og vokser hår..
Hvis sagerne ikke er meget alvorlige, anbefales det at bruge ikke-kirurgiske metoder. Normalt vil disse behandlinger forhindre sygdommens progression, eller de vil blive bedre, men det er normalt, at der fortsat er en vis grad af misdannelse, der kan løses med simpel operation.
Hvis vi er i en situation, hvor der har været andre deformationer i kraniet, såsom positionel plagiocephaly eller en flad side af hovedet på grund af at være i samme position i lang tid på grund af tryk fra livmoderen eller komplikationer i fødslen , kan vi vende tilbage til normal hovedform med en specialformet hjelm til baby.
En anden mulighed er omplacering, som har været effektiv i 80% af tilfældene. Det består i at placere babyen på den upåvirkede side og arbejde på nakkemusklerne ved at placere ham med forsiden nedad på underlivet. Denne teknik er effektiv, hvis babyen er mindre end 3 eller 4 måneder gammel..
Det kirurgiske indgreb, der udføres af en kraniofacial kirurg og en neurokirurg, er indiceret i tilfælde af alvorlige kraniofaciale problemer, såsom lamboid eller coronal craniosynostosis, eller hvis der er en stigning i intrakranielt tryk.
Kirurgi er den valgte behandling for de fleste kraniofaciale misdannelser, især dem, der er forbundet med et større syndrom.
Målet med kirurgi er at mindske det tryk, som kraniet lægger på hjernen og at give tilstrækkelig plads til hjernen til at vokse såvel som at forbedre det fysiske udseende..
Efter operationen har du muligvis brug for en anden intervention senere, hvis du har tendens til at udvikle kraniosynostose, når du bliver ældre. Det samme sker, hvis de også har ansigtsdeformiteter.
En anden type operation er endoskopisk, som er meget mindre invasiv; da det involverer introduktion af et belyst rør (endoskop) gennem små snit i hovedbunden for at opdage den nøjagtige placering af den smeltede sutur for at åbne den senere. Denne type operation kan udføres på så lidt som en time, hævelsen er ikke så alvorlig, der er mindre blodtab, og opsvinget er hurtigere..
I tilfælde af andre underliggende syndromer er periodisk overvågning af kranietvækst nødvendig for at overvåge forekomsten af øget intrakranielt tryk..
Hvis din baby har et underliggende syndrom, kan lægen anbefale regelmæssige opfølgningsbesøg efter operationen for at overvåge hovedvækst og kontrollere for øget intrakranielt tryk..


Endnu ingen kommentarer