Det Duchenne muskeldystrofi (DMD) er en neuromuskulær sygdom, der er kendetegnet ved tilstedeværelsen af signifikant muskelsvaghed og en generaliseret og progressiv udvikling (World Health Organization, 2012).
Det er den mest almindelige type muskeldystrofi hos mennesker (López-Hernández, 2009) og rammer 1 ud af 3.500 børn i verden (Duchenne Parent Project, 2012). For det meste påvirker sygdommen mænd i de tidlige stadier af livet (Verdenssundhedsorganisationen, 2012).
Der er forskellige typer muskeldystrofi. Symptomer begynder typisk i barndommen. Svaghed og tab af muskelmasse forårsager alvorlige vanskeligheder med at erhverve eller opretholde evnen til at gå, trække vejret og / eller sluge (Mayo Clinic, 2013).
Neuromuskulære effekter giver en kronisk prognose. I de fleste tilfælde dør mennesker med Duchenne muskeldystrofi i ung voksenalder på grund af udviklingen af sekundære patologier såsom hjertesvigt eller kardiomyopatier (World Health Organization, 2012).
Artikelindeks
Duchenne muskeldystrofi er en sygdom, der påvirker individet gennem progressiv muskelsvaghed og degeneration (Muscular Dystrophy Association, 2016).
På grund af en genetisk mutation vil fraværet af et specifikt protein hos mennesker med Duchenne muskeldystrofi forårsage tab af muskelfunktionalitet.
Generelt vises symptomerne normalt i underekstremiteterne og spredes til resten af områderne.
Verdenssundhedsorganisationen (2012) indikerer, at forekomsten af Duchenne muskeldystrofi estimeres til ca. 1 tilfælde pr.3.300 indbyggere.
Specifikt viser nogle undersøgelser, at denne sygdom rammer 1 ud af 3.500 mandlige børn født i live (López-Hernández, 2009).
I USA er det ikke kendt med sikkerhed, hvor mange mennesker i alle aldersgrupper, der lider af denne sygdom. Nogle undersøgelser har estimeret, at en ud af 5.600-7.770 mandlige voksne i alderen 5 til 24 år har en diagnose af Duchenne eller Becker muskeldystrofi (Centers for Disease Control and Prevention, 2015).
Det mest karakteristiske ved de lidelser, der hører til gruppen af muskeldystrofi er muskelsvaghed; Afhængigt af typen kan der dog forekomme specifikke symptomer, der varierer afhængigt af debutalderen og de berørte muskelgrupper (Mayo Clinic, 2013).
Normalt er udviklingen af Duchnne muskeldystrofi ret forudsigelig. Forældre kan observere nogle ganske signifikante tegn, såsom vanskeligheder eller manglende evne til at lære at gå eller unormal stigning i lægmuskler (pseudohypertrofi) (Duchenne Parent Project, 2012).
Nogle af de mest karakteristiske symptomer og tegn på Duchenne muskeldystrofi, der vises tidligt i et barns liv, er (Mayo Clinic, 2013):
Tilsvarende fremhæver Duchenne Parent Projet-foreningen (2012) de mest almindelige symptomer og kliniske manifestationer:
Alle muskelsymptomer begynder med svaghed i bækkenbundens muskler, kalve og forskellige gangforstyrrelser, der er signifikante inden 5 år (López-Hernández, 2009).
I børnehaven kan børn med Duchenne muskeldystrofi ofte falde eller have svært ved at gå, klatre trin og / eller løbe (Duchenne Parent Project, 2012).
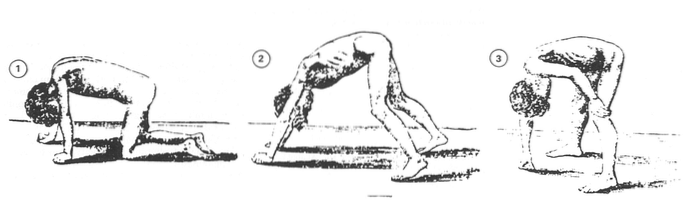
Efterhånden som sygdommen skrider frem, er det meget sandsynligt, at børn kun bruger fodspidserne til at gå i skolealderen. Vi vil være i stand til at observere en rullende og usikker march, der kan forårsage adskillige fald. De bruger normalt nogle strategier til at opretholde deres balance, såsom at skubbe skuldrene tilbage eller holde fast i deres egen krop (Duchenne Parent Project, 2012).
Omkring 9 år er de fleste mennesker med denne sygdom ikke i stand til at gå, på grund af dette begynder de at udvikle adskillige muskuloskeletale deformiteter - skoliose, kontrakturer osv. - (López-Hernández, 2009).
I teenagestadiet vil de præsentere betydelige vanskeligheder ved effektivt at udføre aktiviteter relateret til brugen af de øvre ekstremiteter, ben eller bagagerum. På dette tidspunkt vil de kræve mekanisk støtte og hjælp (Duchenne Parent Project, 2012).
Muskeldegeneration og svaghed fortsætter med at udvikle sig, indtil de når de muskler, der er ansvarlige for åndedræts- og hjertefunktion (López-Hernández, 2009). På grund af alt dette er patientens overlevelse alvorligt kompromitteret og forårsager død i de fleste tilfælde..
Der er identificeret forskellige gener, der er involveret i produktionen af proteiner, der er ansvarlige for at beskytte muskelfibre mod mulig skade og skade (Mayo Clinic, 2013).
Specifikt forekommer hver type muskeldystrofi som en konsekvens af en bestemt genetisk mutation. Nogle af disse mutationer nedarves; dog forekommer de i de fleste tilfælde spontant under graviditet (Mayo Clinic, 2013).
I tilfælde af Duchenne muskeldystrofi identificerede forskerne et specifikt gen placeret på X-kromosomet, der kunne præsentere den mutation, der er ansvarlig for denne patologi (Muscular Dystrophy Association, 2016).
I 1987 blev proteinet, der var forbundet med dette gen, således identificeret., dystrofin. Således indebærer manglen eller fraværet af dette protein, at musklerne er skrøbelige og let beskadiges (Muscular Dystrophy Association, 2016).
Derudover er der identificeret et recessivt arvemønster knyttet til X-kromosomet, hvor bæreren er moderen (Muscular Dystrophy Association, 2016). På grund af denne kendsgerning er denne type sygdom hyppigere hos mænd end hos kvinder..
Mænd har en XY-kromosomsammensætning, mens kvinder er XX. Derfor, hvis et X-kromosom har en mutation i DMD-genet, vil det lide af Duchennes muskeldystrofi på grund af fraværet af dystrofinproduktion (National Human Genome Research Institute, 2013).
I tilfælde af kvinder, der har to X-kromosomer og derfor to kopier af DMD-genet, hvis en af disse ændres, vil den anden være i stand til at fortsætte med at producere dystrophin og derfor opretholde muskelneurobeskyttelse (National Human Genome Research Institute, 2013 ).
I denne type patologier kan forskellige interventioner udføres for at bestemme dens diagnose (National Human Genome Research Institute, 2013).
Den kliniske diagnose kan allerede stilles, når et barn begynder at udvikle progressiv muskelsvaghed. Allerede ved 5 års alderen er der åbenlyse symptomer. Hvis der ikke udføres en tidlig indgriben, vil børn udvise funktionel afhængighed inden 13 år (National Human Genome Research Institute, 2013).
Bortset fra observation og klinisk undersøgelse kan nogle af følgende teknikker bruges til at identificere tilstedeværelsen af Duchenne muskeldystrofi (Mayo Clinic, 2013):
På nuværende tidspunkt er en kur mod Duchennes muskeldystrofi ikke blevet identificeret (Duchenne Parent Project, 2012).
På trods af dette anvendes forskellige behandlinger, der har vist sig at være effektive både til at reducere symptomer og til at forbedre livskvaliteten for mennesker, der lider af denne type patologi (Duchenne Parent Project, 2012).
På grund af den kliniske progression og den brede vifte af symptomer vil denne sygdom kræve en tværfaglig og omfattende intervention udført af en bred vifte af specialister: børnelæge, fysioterapeut, neurolog, neuropsykolog, ergoterapeut, taleterapeut, ernæringsekspert, endokrinolog, genetiker, blandt andet kardiolog, pulmonolog, ortopæd, rehabilitator og kirurg (Duchenne Parent Project, 2012).
I mange tilfælde kan specialister anbefale farmakologiske indgreb (Mayo Clinic, 2013):
Ikke kun er lægemidler nyttige til intervention i Duchennes muskeldystrofi, der er både terapeutiske interventioner og plejemetoder, der kan forbedre livskvaliteten for disse mennesker (Mayo Clinic, 2013).
Nogle gavnlige indgreb er (Duchenne Parent Project, 2012):
Indtil for relativt få år siden overlevede mennesker med Duchenne muskeldystrofi ikke meget længere efter at have nået ungdomsårene (Muscular Dystrophy Association, 2016).
De store fremskridt inden for medicinsk, teknisk og genetisk forskning har formået både at bremse sygdommens progression og at give en betydelig stigning i livskvaliteten til personer, der lider af den (Muscular Dystrophy Association, 2016). På denne måde er hjerte- og åndedrætspleje afgørende for bevarelsen af vitale funktioner (Muscular Distrophy Association, 2016).
I mange tilfælde er de i stand til at nå post-adolescent stadier. Flere og flere tilfælde af Duchenne muskeldystrofi beskrives hos voksne i 30'erne, herunder mennesker, der overlever til 40'erne og 50'erne (Muscular Dystrophy Associatin, 2016).
I øjeblikket er kliniske forsøg og forskning orienteret mod udvikling af genterapier, der ændrer mutationer og mangler i dystrofinproduktion (Muscular Dystrophy Association, 2016).
Nogle af de mest undersøgte mekanismer er (López-Hernández, 2009):
Duchennes muskeldystrofi er en alvorligt invaliderende sygdom hos både børn og unge voksne med en ødelæggende prognose..
På trods af at klinisk og eksperimentel forskning har gjort vigtige fremskridt i behandlingen af symptomer, er der stadig ingen kur mod denne type patologi..
En grundig forståelse af det biologiske og genetiske grundlag er afgørende for at finde en helbredende behandling for Duchennes muskeldystrofi..



Endnu ingen kommentarer