
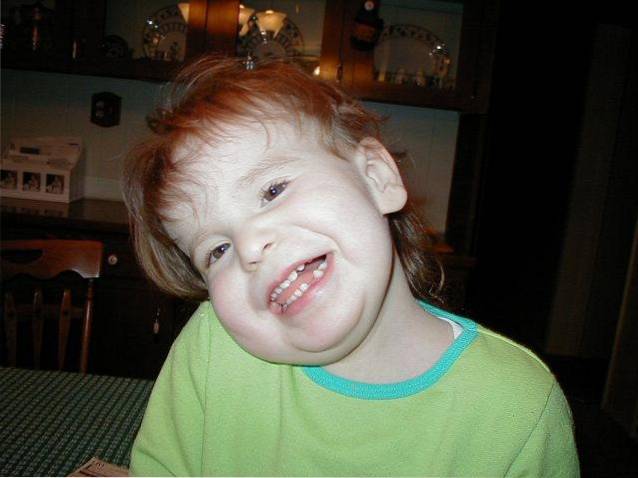
Det Pallister-Killian syndrom, også kendt som tetrasomi 12, er det en sjælden sygdom af genetisk oprindelse, der er kendetegnet ved et bredt spektrum af multi-organ involvering.
Klinisk er denne patologi defineret af intellektuel handicap, psykomotorisk retardering, muskulær hypotoni, en atypisk ansigtsfænotype, pigmentabnormiteter i huden og alopeci. Derudover kan andre typer medicinske komplikationer relateret til misdannelser i forskellige kropssystemer eller anfald også forekomme..
Den etiologiske oprindelse af denne sygdom er forbundet med en genetisk lidelse fordelt i mosaik. Specifikt skyldes det tilstedeværelsen af et ekstra kromosom 12 i nogle celler i kroppen.
Diagnosen af Pallister-Killiam syndrom kan stilles både prænatalt og postnatalt. Hovedformålet er identifikation af de kliniske egenskaber og brugen af en bekræftende genetisk undersøgelse..
Dette syndrom har en høj dødelighed. Imidlertid kan den farmakologiske medicinske tilgang og rehabiliterende behandling give vigtige fordele i livskvaliteten og den kliniske status for de berørte.
Artikelindeks
Denne sygdom blev oprindeligt beskrevet af Pallister i 1977. I de første publikationer rapporterede denne forsker to tilfælde af voksne patienter, hvis forløb var præget af forskellige fund: anfald, muskulær hypotoni, intellektuelt underskud, muskuloskeletale og organiske misdannelser, konfiguration ru ansigts- og hudfarve ændringer.
Parallelt beskrev Teschler-Nicola og Killiam i 1981 det samme kliniske billede i en treårig pige.
Derfor blev der i de første kliniske rapporter en generel henvisning til en medicinsk tilstand karakteriseret ved kombinationen af anfald, intellektuel handicap og en karakteristisk fysisk fænotype..
Derudover var Gilgenkratz allerede i 1985 i stand til i første omgang at identificere under drægtighedsfasen, noget der er almindeligt i dag takket være moderne diagnostiske teknikker.
Pallister-Killiam syndrom er en type genetisk mosaiksygdom. I dette tilfælde påvirker den kromosomale ændring kun nogle celler i kroppen. En bred involvering af forskellige kropssystemer og organismer identificeres.
Det er hovedsageligt præget af intellektuel handicap, muskuløs hypotoni, udvikling af karakteristiske ansigtsegenskaber, ændring af hudpigmentering eller hårvækst, blandt andre medfødte ændringer..
Derudover er Pallister-Kiliam syndrom en sjælden sygdom med medfødt oprindelse, der kan modtage et stort antal navne i den medicinske litteratur:
Prevalenstal for Pallister-Killiam syndrom er ikke nøjagtigt kendt. Der er ikke stillet mange endelige diagnoser, og de fleste af disse er ikke blevet offentliggjort i den medicinske litteratur.
Således definerer alle forfattere og institutioner dette syndrom som en sjælden eller sjælden genetisk patologi i den almindelige befolkning..
For omkring 15 år siden var Pallister-Killiam syndrom blevet identificeret i næsten 100 tilfælde over hele verden. I øjeblikket har dette tal oversteget 200 berørte.
Epidemiologiske undersøgelser har estimeret forekomsten af denne sygdom til ca. 5,1 tilfælde pr. Million nyfødte børn, selvom forfattere som Toledo-Bravo de la Laguna og samarbejdspartnere placerer den på 1 / 25.000.
En højere prævalens forbundet med de sociodemografiske egenskaber hos de berørte er ikke identificeret. Pallister-Killian syndrom kan forekomme i ethvert køn eller teknisk og / eller race gruppe.
En lang række tegn og symptomer kan identificeres i det kliniske forløb af Pallister-Killian syndrom. Alle sammen forbundet med kraniofaciale og / eller muskuloskeletale abnormiteter og kognitive ændringer.
Udviklingen af kranio-ansigtsmisdannelser fra drægtighedsfasen til postnatal og spædbarnsvækst udgør et af de mest karakteristiske medicinske tegn på Pallister-Killiam syndrom..
De mest almindelige tegn og symptomer inkluderer abnormiteter i de forskellige kranie- og ansigtsstrukturer, der vil føre til et ru og atypisk udseende:
På trods af at det er mindre signifikant end ansigtsændringer, er det meget almindeligt at observere flere muskuloskeletale abnormiteter hos patienter, der er ramt af Pallister syndrom:
Abnormiteter relateret til muskelstruktur og mobilitet er et andet af de kardinal kliniske træk ved Pallister-Killian syndrom:
Muskelhypotoni refererer til identifikation af unormalt reduceret muskeltonus eller spænding. På et visuelt niveau kan slaphed og labilitet observeres i forskellige muskelgrupper, især forstærket i ekstremiteterne..
Muskel- og skeletpatologi vil således medføre en betydelig forsinkelse i erhvervelsen af forskellige motoriske færdigheder, både i nyfødte og barndomsperioder..
Selvom udviklingsperioderne er varierende blandt de berørte, inkluderer den mest almindelige kalender følgende milepæle:
Et andet af de stærkt berørte områder er nervesystemet. I de fleste tilfælde er tegn og symptomer hovedsageligt relateret til anfald og intellektuel handicap:
Selvom de er mindre hyppige, kan andre typer medicinske komplikationer også forekomme:
Oprindelsen til Pallister-Killian syndrom er forbundet med en genetisk mosaik abnormitet på kromosom 12. Det påvirker kun det genetiske materiale i nogle celler i kroppen..
Kromosomer er en del af kernen i alle celler, der findes i menneskekroppen. De består af en lang række biokemiske komponenter og indeholder de enkelte menneskers genetiske information..
Mennesker har 46 forskellige kromosomer, organiseret i par og nummereret fra 1 til 23. Derudover har hvert kromosom på det individuelle niveau et kort område eller en arm kaldet "p" og en lang kaldet "q".
Abnormaliteten påvirker kromosom 12 og resulterer i tilstedeværelsen af et kromosom med en unormal struktur, kaldet et isochromosom..
Således har dette kromosom tendens til at have to korte arme i stedet for en af hver p (kort) og lang (q) konfiguration..
Som en konsekvens vil tilstedeværelsen af ekstra og / eller unormalt genetisk materiale ændre det normale og effektive forløb af den berørte persons fysiske og kognitive udvikling, hvilket giver anledning til de kliniske egenskaber ved Pallister-Killian syndrom..
Pallister-Killian syndrom kan identificeres under graviditet eller i postnatalt stadium baseret på de kliniske egenskaber og resultaterne af forskellige laboratorietests..
Under graviditet er de mest anvendte tests ultralydsscanninger, fostervandsprøve eller chorionisk villusprøveudtagning. I denne forstand kan analysen af embryonets genetiske materiale give os en bekræftelse af denne patologi gennem identifikation af kompatible uregelmæssigheder..
På den anden side, hvis diagnosen stilles efter fødslen, er det vigtigt:
Ingen specifikke terapier er designet til behandling af mennesker med Pallister-Killian syndrom..
Dette syndrom er normalt forbundet med en dårlig neurologisk prognose og høje dødelighedstal. Rehabiliterende behandling, specialundervisning og ergoterapi kan dog tilbyde en god funktionel prognose og en forøgelse af livskvaliteten for de berørte..
For eksempel beskriver Méndez og hans arbejdsteam (2013) et tilfælde af rehabiliterende behandling præget af:

Endnu ingen kommentarer