Det prioner de er proteiner uden et genom eller nukleinsyrer, der fungerer som infektiøse stoffer. Udtrykket "prion" betyder proteinholdig infektiøs partikel (fra engelske proteinholdige infektiøse partikler) og blev opfundet af neurologen og nobelprisvinderen, Stanley B. Prusiner.
I 1982 identificerede Prusiner og hans kolleger en infektiøs proteinpartikel, mens de studerede årsagerne til Creutzfeldt-Jakobs sygdomme (hos mennesker) og bovin spongiform encefalopati..
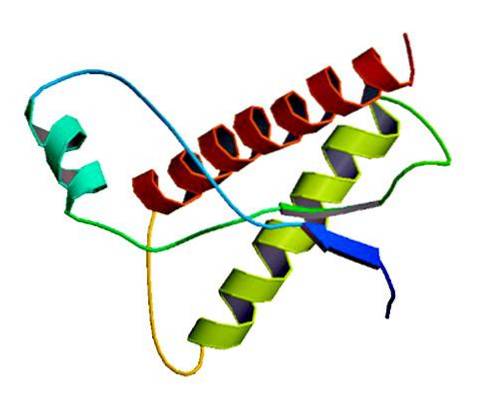
Disse sjældne infektiøse stoffer findes i normale celler i membranen, kun som misfoldede proteiner og / eller med en unormal tredimensionel struktur. Disse proteiner er ansvarlige for flere degenerative sygdomme og meget høj dødelighed, der påvirker neurale væv og hjernens struktur..
De kaldes også prionsygdomme. Blandt de vigtigste, der påvirker mennesker, er kuru, Gerstmann-Sträussler-Scheinker sygdom, Creutzfeldt-Jakob syndrom og dødelig familiær søvnløshed..
Artikelindeks
Prioner er proteinstrukturer, der findes i cellemembraner. Disse proteiner har en ændret form eller konformation [PrP (Sc)].
Med hensyn til dens multiplikation opnås det ved konvertering af former, som i tilfælde af scrapiesygdom. I denne sygdom rekrutterer prioner PrP (C) (prionproteiner med uændret konformation) for at stimulere omdannelse til PrP (Sc) isoformen..
Dette genererer en kædereaktion, der spreder det infektiøse materiale og muliggør derfor vanding af sygdommen. Det vides stadig ikke, hvordan denne konverteringsproces opstår.
Disse usædvanlige proteiner, der er i stand til formering, har ikke nukleinsyrer. Bevis for dette er, at de er modstandsdygtige over for røntgenstråler og ultraviolet stråling. Disse stoffer nedbryder let nukleinsyrer.
Prionproteiner, hvoraf prioner (PrP) er sammensat, findes i hele kroppen, ikke kun hos mennesker, men i andre sunde hvirveldyr. Disse proteiner er generelt resistente over for proteaser (enzymer, der katalyserer proteiner).
Meget lidt er kendt om anvendeligheden af prionproteinerne PrP (C), den normale form for det ikke-infektiøse protein i den menneskelige krop..
Imidlertid har nogle forskere lykkedes at vise, at disse proteiner hos mus aktiverer myelinreparation i celler i det perifere nervesystem. Fraværet af disse har også vist sig at forårsage demyelinering af sådanne nerveceller..
Den viden, der er haft om strukturen af prioner, ligger hovedsageligt i de undersøgelser, der er udført i bakterien Escherichia coli.
Undersøgelser har vist, at kædepolypeptiderne PrP (C) (normal) og PrP (Sc) (infektiøs) er identiske med hensyn til aminosyresammensætning, men adskiller sig i 3D-konformation og foldning..
Disse ikke-infektiøse prioner har 209 aminosyrer hos mennesker. De har en disulfidbinding. Dens struktur er alfa-spiralformet, hvilket betyder, at den har spiralformede aminosyrer (alpha helices) og få flade tråde af aminosyrer (beta-ark)..
Dette protein kan ikke adskilles ved centrifugering, hvilket indebærer, at det ikke er sedimenterbart. Det fordøjes let af bredspektret serinprotease kaldet proteinase K.
Det er et infektiøst protein, der transformerer PrP (C) til infektiøs PrP (Sc) isoformer og med en unormal konfiguration eller form.
Meget lidt er kendt om dens 3D-struktur, men det vides, at den har få spiralformede former og flere flade tråde eller beta-ark. Skiftet til isoformen er det, der er kendt som den centrale begivenhed af prionsygdomme.
Cellulære prionproteiner [Prp (C)] er placeret på celleoverfladen i en lang række organer og væv. Meget lidt er kendt om de fysiologiske funktioner i prioner i kroppen. Alligevel indikerer eksperimenter udført på mus mulige funktioner, såsom:
PrP (C) har vist sig at virke med glutamatreceptorer (ionotrope og metabotrope). PrP (C) deltager som en receptor for synaptotoksiske oligomerer af celleoverfladepeptidet Ap.
Hos mus af Murinae-familien er det blevet opdaget, at prionproteinerne PrP (C) udtrykkes inden for få dage efter implantation i embryonal udvikling.
Dette indikerer, at de spiller en rolle under udviklingen af disse små pattedyr. Den rolle, der ifølge forskerne er relateret til reguleringen af neuritogenese (produktion af axoner og dendritter af neuroner).
De virker også på aksonal vækst. Disse prionproteiner er endda involveret i udviklingen af cerebellar kredsløb. På grund af dette antages det, at fraværet af disse PrP (C) -prioner medfører en forsinkelse i motorudviklingen af gnavere..
I undersøgelser af overekspression af PrP (C) ved genorientering blev det fundet, at fraværet af disse prioner forårsager problemer med blodtilførslen til nogle dele af hjernen (akut cerebral iskæmi).
Dette betyder, at prionproteiner fungerer som neurobeskyttere. Derudover er det vist, at PrP (C) overekspression kan reducere eller forbedre skader forårsaget af iskæmi..
Den fysiologiske rolle Prp (C) i vedligeholdelsen af perifert myelin blev for nylig opdaget.
Under en laboratorieundersøgelse blev det opdaget, at laboratoriemus i mangel af prionprotein udviklede mangler i nerverne, der bærer information fra hjernen og rygmarven, i det, der kaldes perifer neuropati..
Der er nogle proteiner, der ligner prioner, og disse er placeret i andre dele af kroppen end hjernen.
Funktionerne af sådanne proteiner er at initiere, regulere og / eller kontrollere celledød, når organismen bliver angrebet (f.eks. Af vironer) og således forhindrer spredning af patogenet..
Denne ejendommelige funktion af disse proteiner får forskere til at tænke over den mulige betydning af ikke-infektiøse prioner i kampen mod patogener..
En undersøgelse udført ved Stowers Institute i Missouri, USA viste, at PrP-prioner kan have en rolle i at opretholde langvarig hukommelse.
Undersøgelsen afslørede, at visse prionproteiner kan styres til at arbejde med at bevare de fysiologiske funktioner i langtidshukommelsen..
En undersøgelse af prionproteiner, der udtrykkes i stamceller i blodvæv, afslørede, at alle disse stamceller (hæmatopoietiske) udtrykker prionproteiner i deres cellemembran. For hvad antages det, at de deltager i den komplekse og meget vigtige proces med cellefornyelse.
Patologier af prion-oprindelse anerkendes som progressive degenerative hjernesygdomme. De kan angribe kvæg, hjorte, rensdyr, får og endda mennesker.
Disse sygdomme er forårsaget af en ændring i strukturen af PrP (C) proteiner, og hvis specifikke funktioner stadig er usikre i dag. Prionpatologier kan opstå uden en kendt årsag. De kan have en arvelig genetisk oprindelse og kan også overføres på en smitsom-smitsom måde.
Prioner forårsager familiære, sporadiske og smitsomme sygdomme. Familiale prionsygdomme er de, der er arvelige. Sporadiske patologier er de mest almindelige og forekommer uden kendte årsager..
Smitsomme sygdomme betragtes som sjældne, de overføres af person til person, dyr til dyr, person til dyr og omvendt. Årsagerne er flere og spænder fra forbrug af kontamineret kød, kannibalisme, transfusioner til manipulation af kontamineret kirurgisk udstyr.
De mest almindelige prionsygdomme er:
Betragtes som den mest almindelige prionsygdom blandt mennesker, det er en kosmopolitisk sygdom, det vil sige den har en verdensomspændende fordeling. Det kan være arveligt (familiært), sporadisk eller infektiøst.
Patienter med symptomer som demens, ryk eller pludselige ufrivillige bevægelser og mangler i centralnervesystemet.
Afhængig af behandlingen og formen af sygdommen kan døden forekomme mellem 4 måneder og 2 år efter erhvervelsen af sygdommen. Diagnose er vanskelig at stille, det gøres normalt post morten, under obduktion.
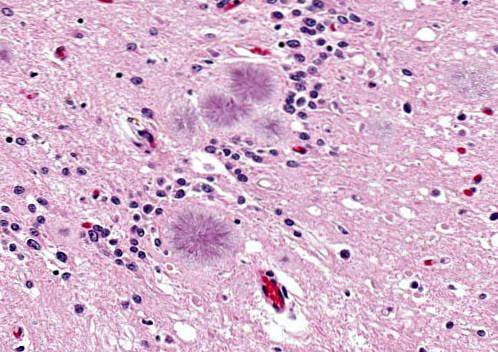
Det er en sygdom forårsaget af prioner i en arvelig eller autosomal dominerende infektiøs hjerneproces. Sygdommen manifesterer sig hos mennesker i alderen 40 til 60 år.
Disse mennesker manifesterer problemer med at artikulere ord (dysartri), ryk eller pludselige ufrivillige bevægelser, aggressivitet er hyppig.
De præsenteres med cerebellær degeneration ledsaget af en ustabil gangart. Det er også muligt at observere hyporefleksi, døvhed, bliklammelse, demens, blandt andre symptomer. Forventet levetid er cirka 5 år eller lidt længere.
Det er en meget sjælden sygdom, til det punkt, at dens forekomstsområde er 2 til 3 tilfælde pr. 100 millioner indbyggere. Patologien svarer til Gerstmann-Sträussler-Scheinkers sygdom.
Proteinets kliniske manifestationer indikerer lav resistens over for proteaser, nogle er mere og andre mindre følsomme over for disse enzymer.
De symptomer, som patienter præsenterer, er: problemer med tale og kognitiv svækkelse, tab af neuroner i det område, hvor hjernen styrer bevægelser og udfører muskelkoordinering.
Sygdommen er hyppig hos ældre patienter (70 år), og den estimerede levetid, når den først er smittet, er ca. 20 måneder.
Det er en arvelig eller familiær sygdom, den kan også forekomme sporadisk. Sygdommen vides at være på grund af en arvelig eller autosomal dominerende mutation.
Patienter præsenterer symptomer som kumulative problemer med at sove og vedligeholde søvn, demens, kognitiv svækkelse, endda problemer med hypertension, takykardi, hyperhidrose og andre..
Den alder, det påvirker, er ret bred og spænder mellem 23 og 73 år, men gennemsnitsalderen er 40 år. Levetiden, når den først er smittet, er lidt over 6 år.
Denne prionsygdom er kun påvist hos indbyggerne i Papua Ny Guinea. Det er en sygdom relateret til kannibalisme og den kulturelle tradition for riten om at sørge over de døde, hvor disse mennesker spiser hjerne eller menneskekød.
Mennesker, der bærer sygdommen, har normalt ukontrollerbare og ufrivillige bevægelser i forskellige dele af kroppen.
De præsenterer rystelser, tab af kontrol over bevægelser og tab af muskelkoordination. Forventet levetid hos inficerede mennesker er to år.
Blandt patologier produceret af prioner hos dyr er bovin spongiform encefalopati. Denne sygdom forårsagede kaos i Europa, folkesundheden, hos dyr og i de berørte landes økonomi.
Andre sygdomme hos dyr inkluderer scrapie, overførbar minkeencefalopati, kronisk spildsygdom (hos hjorte) og feline spongiform encefalopati..
Disse sygdomme mangler, ligesom dem, der præsenteres hos mennesker, en effektiv behandling, så forebyggelse er vigtig, især efter infektioner hos mennesker, der er opstået som et resultat af forbruget af kød fra inficerede køer..
Til dato er der ingen kendt kur mod prionsygdomme. Behandling er symptomatisk. Patienter rådes til at planlægge palliativ behandling og genetisk testning og rådgivning til familiemedlemmer anbefales.
En bred vifte af medikamenter er blevet testet hos patienter med prionsygdomme, såsom antivirale midler, antitumorer, lægemidler mod sygdomme såsom Parkinsons, behandlinger mod immunsuppression, antibiotika, svampedræbende midler, endda antidepressiva..
Der er dog i øjeblikket ingen beviser for, at nogle af disse reducerer symptomerne eller forbedrer patienternes overlevelse..
Prioner er resistente over for en række fysiske og kemiske ændringer. Imidlertid anvendes forskellige teknikker til at undgå kontaminering af patienter med kontaminerede kirurgiske instrumenter..
Blandt de mest anvendte teknikker er at sterilisere udstyret i en autoklav ved 132 ° C i en time og derefter nedsænke instrumenterne i natriumhydroxid i mindst en time mere..
På den anden side har verdenssundhedsorganisationen (WHO) udviklet foranstaltninger til at forhindre spredning af prionsygdomme. Denne organisation fastlægger normer for håndtering af forbudte eller potentielt risikable væv såsom: øjne, hjerne, tarm, mandler og rygmarv.



Endnu ingen kommentarer