Det robinow syndrom det er en patologi af sjælden genetisk oprindelse, der er kendetegnet ved tilstedeværelsen af flere ændringer og kropsmisdannelser, især på knogleniveau.
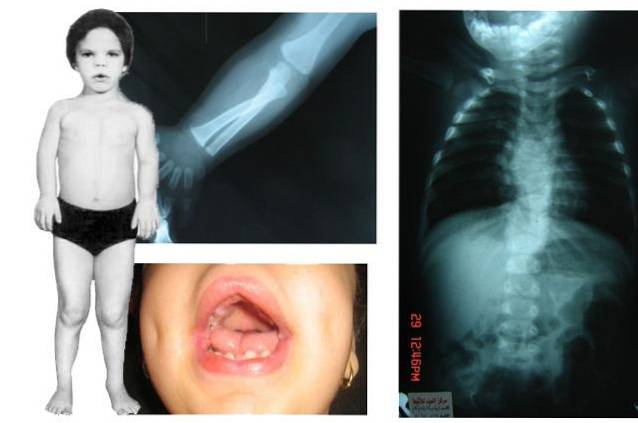
På et klinisk niveau er det en sygdom, der blandt andet kan påvirke forskellige områder såsom kraniofacial, muskuloskeletale, orale og urogenitale strukturer. Derudover inkluderer nogle af de hyppigste tegn og symptomer i denne patologi: makrocephali, kort statur, kønshypoplasi og atypiske ansigtsegenskaber, blandt andre..
Med hensyn til etiologien af Robinow syndrom er det i øjeblikket forbundet med tilstedeværelsen af specifikke mutationer i ROR2-, WNT5A-, DVL1-generne, der er til stede differentielt afhængigt af det specifikke arvelighedsmønster i hvert tilfælde.
Der er ingen specifikke tests eller biologiske markører, der specifikt indikerer tilstedeværelsen af Robinow syndrom, af denne grund er diagnosen baseret på undersøgelse af det kliniske billede og den radiologiske undersøgelse.
Robinow syndrom er til stede fra fødslen, så en kur er endnu ikke identificeret; behandlingen er hovedsagelig symptomatisk, den fokuserer på kontrol af medicinske komplikationer, såsom åndedræts- eller hjerteforandringer.
Artikelindeks
Robinow syndrom er en sygdom af arvelig oprindelse, hvis centrale kendetegn er den generelle forsinkelse i fysisk udvikling, hvilket giver anledning til tilstedeværelse af kort eller reduceret statur, kranioaciale misdannelser og andre ændringer i bevægeapparatet.
Denne patologi blev oprindeligt beskrevet i 1969 af Minhar Robinow. I sin kliniske rapport beskrev han en række tilfælde karakteriseret ved unormale eller atypiske ansigtsegenskaber, kort statur eller hypoplastiske kønsorganer, hvis etiologiske oprindelse var autosomalt dominerende..
Imidlertid viste efterfølgende undersøgelser gennem de gennemgåede sager, at Robinow syndrom er en meget heterogen patologi, så dets kliniske og morfologiske træk kan variere betydeligt på tværs af forskellige tilfælde..
Derudover er denne sygdom også kendt som føtal facies syndrom, Robinows dværgisme, Robinows mesomeliske dysplasi eller dysotosis acra med ansigts- og kønsforstyrrelser.
Generelt er den medicinske prognose for Robinow syndrom god, da forventet levealder ikke er reduceret i forhold til den generelle befolkning, men den har en høj grad af comorbiditet, så livskvaliteten påvirkes betydeligt.
Robinow syndrom er sjældent i hele verden, hvorfor det betragtes som en sjælden sygdom.
Specifikt er ca. 200 tilfælde af Robinow-syndrom med en autosomal recessiv arvelig oprindelse beskrevet i den medicinske litteratur, mens den dominerende form er blevet identificeret i mindst 50 familier..
På den anden side er forekomsten af Robinow-syndrom estimeret til ca. 1-6 tilfælde pr. 500.000 fødsler hvert år..
Derudover har det ikke været muligt at identificere en forskellig hyppighed med hensyn til køn, geografisk oprindelse eller etniske og racemæssige grupper, selvom klinisk identifikation i nogle tilfælde er hurtigere hos mænd på grund af kønsanomalier.
Mønstret for involvering af Robinow syndrom er bredt, da det påvirker hele kropsstrukturen på en generaliseret måde og især kraniofaciale, bukkale, kønsorganer og muskuloskeletale områder.
Nogle af de hyppigere ændringer inkluderer:
Mennesker, der lider af Robinow-syndrom, har en alvorlig påvirkning af kranie- og ansigtsstrukturen, hvilket giver dem en atypisk konfiguration og udseende. Nogle af de mere almindelige abnormiteter inkluderer:
- Kraniale abnormiteter: det mest almindelige er at observere et kranialvolumen, der er større end forventet for dets udviklingsmoment (makrocephali), ledsaget af en udbulende frontal fremtrædende eller pande og dårlig eller ufuldstændig udvikling af de nedre dele af ansigtet (ansigtshypoplasi).
- Okulær hypertelorisme: dette udtryk refererer til tilstedeværelsen af en unormal eller overdreven adskillelse af de okulære baner. Derudover er udviklingen af unormalt fremtrædende øjne med tilbøjelighed til de palpebrale revner almindelig..
- Næse abnormiteter: næsen præsenterer normalt en reduceret eller forkortet struktur ledsaget af en kløft i næsen eller ændringer i dens position.
- Strukturelle orale abnormiteter: i tilfælde af munden er det almindeligt at observere en trekantet struktur ledsaget af en lille kæbe (micrognathia).
Disse typer af ændringer henviser til en mangelfuld eller unormal organisering af mundens indre struktur og tandorganisationen..
- Dental ændringer- Tænderne er ofte forkert justeret med bageste gruppering eller forsinket udbrud af sekundære tænder.
- Gingival hyperplasi: både tyggegummi såvel som resten af blødt væv og mundstrukturer kan vise et forstørret eller betændt udseende.
På muskuloskeletalt niveau udgør knoglemedvirkning et af de mest betydningsfulde medicinske symptomer i Robinow syndrom.
- Kort statur: fra graviditet eller fødselsøjeblikket er det muligt at opdage en forsinket fysisk udvikling, knoglealderen er normalt lavere end den kronologiske alder, så andre aspekter påvirkes, såsom højde, som normalt reduceres og ikke når forventede standarder.
- Vertebrale ændringer: rygsøjlens knoglestruktur har tendens til at have en dårlig organisation, det er muligt, at en underudvikling af vertebrale knogler eller en fusion af en af dem vises. Derudover er tilstedeværelsen af skoliose eller en unormal og patologisk krumning af rygsøjlen også meget almindelig..
- Brachymellia: knoglerne, der bekræfter armene, forkorteres normalt i længden, så armene ser kortere ud end normalt.
- Kinodactyly: der er en lateral afvigelse af nogle fingre i hånden, der især påvirker tommelfingeren og / eller ringfingeren.
Kønsmæssige abnormiteter er også almindelige hos børn med Rainbow-syndrom, og de er især tydelige hos drenge..
- Genital hypoplasi: generelt er kønsorganerne ikke fuldt udviklede, det er især almindeligt at observere tvetydige kønsorganer, der er dårligt differentieret som mand eller kvinde.
- Cryptorchidism: i tilfælde af mænd kan kønsudvikling forårsage delvis eller fuldstændig fravær af nedstigningen af testiklerne mod pungen.
- Nyresygdomme: nyrefunktion er også normalt påvirket, da hyppig lider af hydronephrose (ophobning af urin i nyrerne).
Ud over de abnormiteter, der er beskrevet ovenfor, er det meget almindeligt at observere udviklingen af hjerteabnormiteter og abnormiteter. De mest almindelige er relateret til obstruktion af blodgennemstrømningen på grund af strukturelle misdannelser.
På den anden side findes der i det neurologiske område normalt ikke signifikante træk, da intelligens præsenterer et standardniveau såvel som kognitive funktioner. Kun i nogle tilfælde er det muligt at observere en lille forsinkelse.
Robinow syndrom er en medfødt arvelig sygdom, så den har en klar genetisk etiologisk karakter.
På trods af at forskellige genetiske komponenter relateret til det kliniske forløb af Robinow syndrom er blevet identificeret, specifikt ROR2-, WNT5A- og DVL1-generne, er det arvelige mønster endnu ikke kendt nøjagtigt, det er også differentieret er mange berørt.
Specifikt synes tilfælde af Robinow-syndrom, der er forbundet med specifikke mutationer af ROR2-genet, placeret på kromosom 9 (9q22), at præsentere et autosomalt recessivt mønster af arvelighed.
I tilfælde af recessive genetiske patologier er det nødvendigt at have i det individuelle genetiske materiale to kopier af det unormale eller defekte gen, der kommer fra begge forældre, en fra hver af dem.
Men hvis personen kun arver en af disse, vil de være en bærer, dvs. de vil ikke udvikle de kliniske egenskaber ved Robinow syndrom, men de vil være i stand til at overføre det til deres afkom.
I dette tilfælde har ROR2-genet den væsentlige funktion til at generere de essentielle biokemiske instruktioner til produktion af et protein, der er vigtig for normal fysisk udvikling under prænatalstadiet. Specifikt er ROR2-proteinet vigtigt for dannelsen af kroppens knoglestruktur, hjertet og kønsorganerne.
Som et resultat vil tilstedeværelsen af genetiske ændringer, der påvirker den effektive funktion af denne komponent, afbryde normaliseret fysisk udvikling og vises derfor som de karakteristiske kliniske egenskaber ved Robinow-syndrom..
Imidlertid er de dominerende former for Robinow-syndrom forbundet med tilstedeværelsen af specifikke mutationer i WNT5- eller DVL1-genet..
I tilfælde af genetiske patologier af dominerende oprindelse kan deres kliniske forløb udvikle sig fra en enkelt defekt genkopi fra en af forældrene eller fra udviklingen af en ny mutation.
Specifikt synes de proteiner, der genererer WNT5- og DVL1-generne, at være involveret i det samme funktionelle mønster som ROR2'erne, så tilstedeværelsen af abnormiteter og mutationer i dem ændrer signalveje, der er væsentlige for fysisk udvikling..
Diagnosen af Robinow syndrom er grundlæggende klinisk, derfor er den baseret på observation af det kliniske forløb, studiet af individets og familiens medicinske historie og den fysiske undersøgelse.
Nogle fund skal bekræftes gennem radiologiske tests, især knoglemangel (ekstremiteter, kranium, rygsøjle osv.).
Ud over diagnosen under det infantile eller nyfødte stadium er det også muligt at bekræfte det under graviditeten. Undersøgelsen af længden af forskellige knoglekomponenter er især indiceret i føtal ultralyd i tilfælde af genetisk risiko.
På den anden side udføres i begge tilfælde normalt en genetisk undersøgelse for at analysere den mulige tilstedeværelse af genetiske mutationer, der forklarer oprindelsen til Robinow syndrom..
Derudover er det vigtigt at udføre den differentielle diagnose med andre typer patologier, der har lignende kliniske træk, især tilstedeværelsen af atypiske ansigtsegenskaber. Således er de vigtigste patologier, der er udelukket, hypertelorisme, Aarskog-Scott syndrom eller Opitz syndrom..
Der er i øjeblikket ingen kur mod Robinow syndrom, så den terapeutiske behandling af denne sygdom fokuserer på opløsning af medicinske komplikationer..
Muskuloskeletale lidelser løses normalt gennem fysioterapi, proteseplacering eller korrektion gennem kirurgiske procedurer. På den anden side behandles hjerte- og kønsændringer normalt gennem farmakologiske og / eller kirurgiske behandlinger..
Derudover er der også andre typer nye terapier, der er baseret på administration af væksthormoner for at stimulere stigningen i højden. Det kan dog have forskellige bivirkninger, såsom forværring af skoliose..
Sammenfattende er tidlig terapeutisk intervention vigtig for korrektion af muskuloskeletale ændringer og kontrol af medicinske komplikationer, såsom hjerte manifestationer..
Ligeledes er arbejdet i tværfaglige teams, fysisk, social og psykologisk intervention, afgørende for at fremme udviklingen af kapaciteter og evner hos berørte børn.
På denne måde er formålet med interventionen at lade den berørte person nå sit maksimale udviklingspotentiale, opnå funktionel afhængighed og en optimal livskvalitet..

Endnu ingen kommentarer